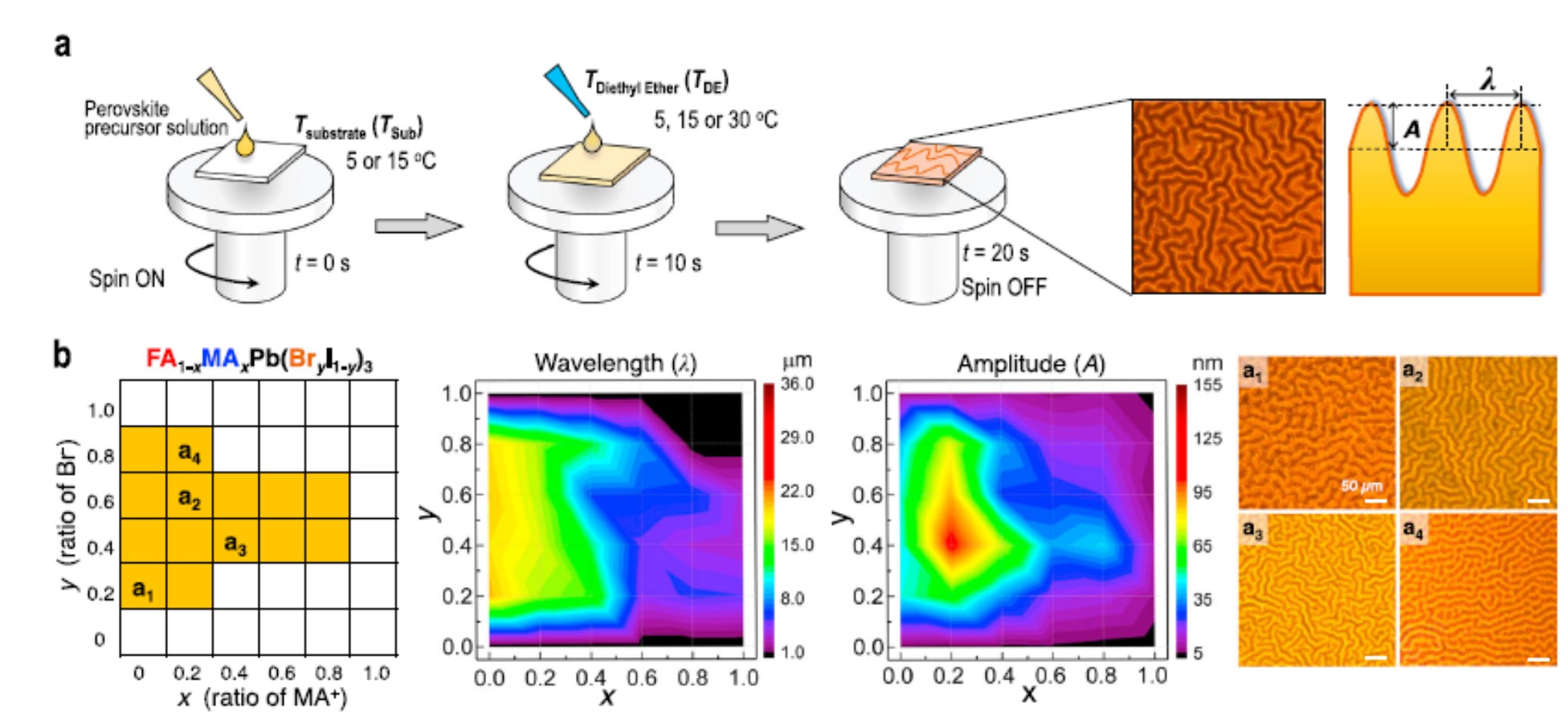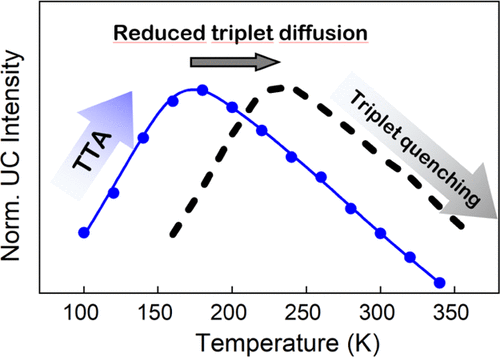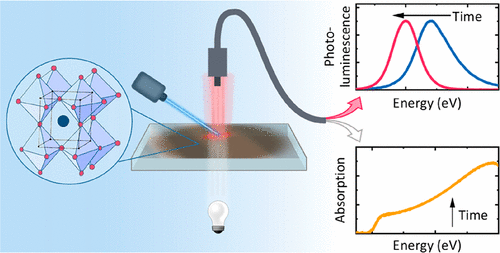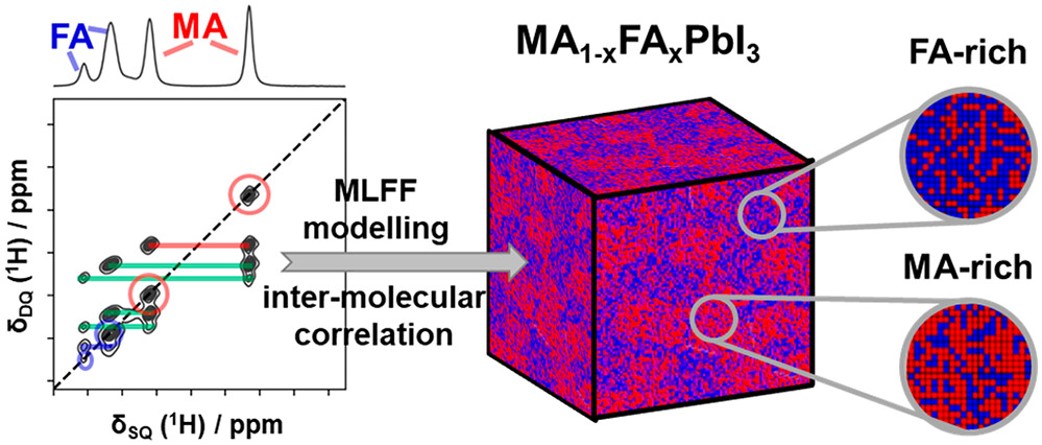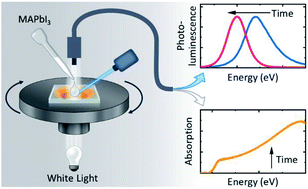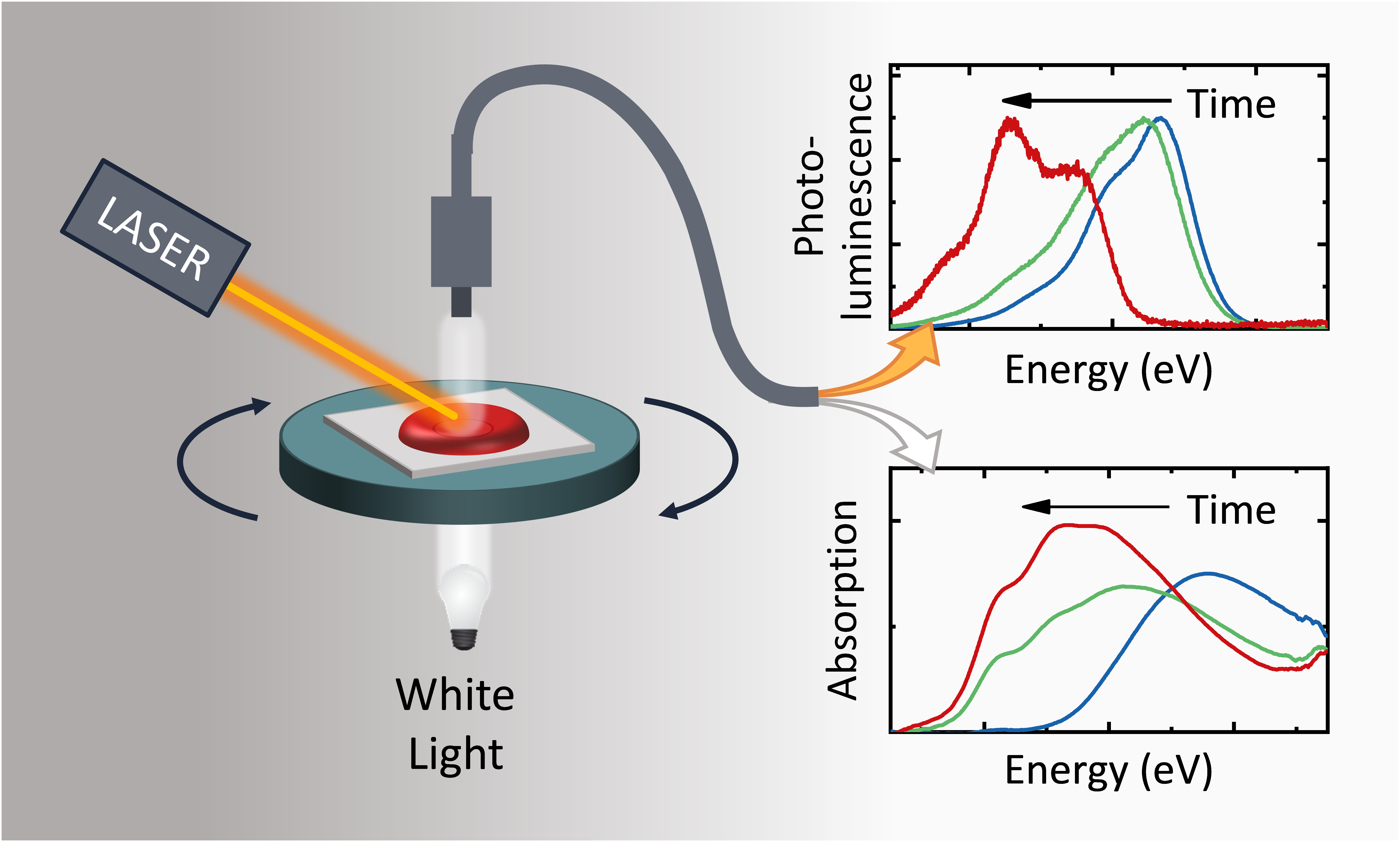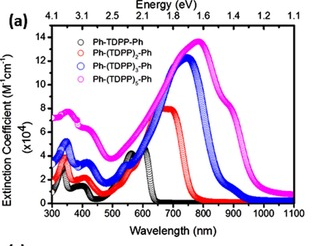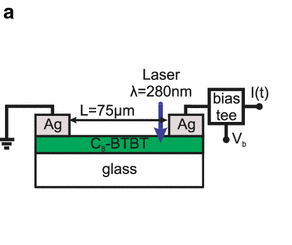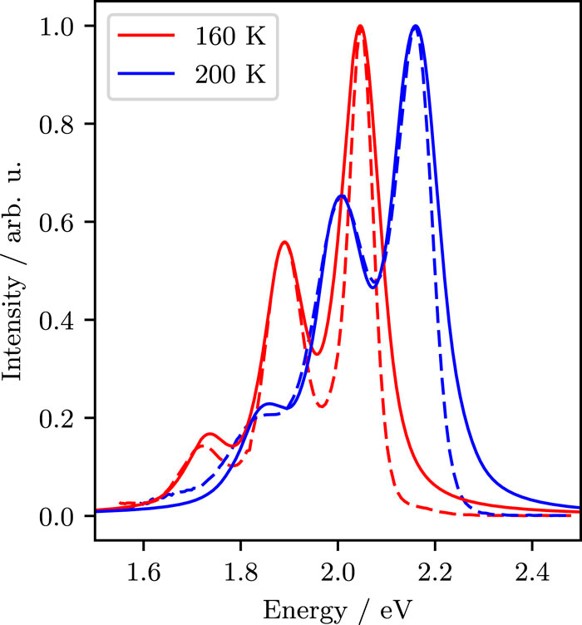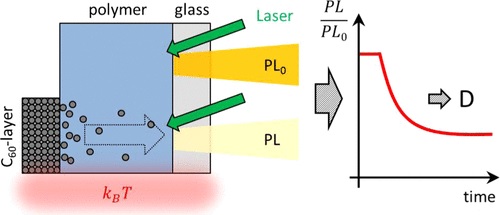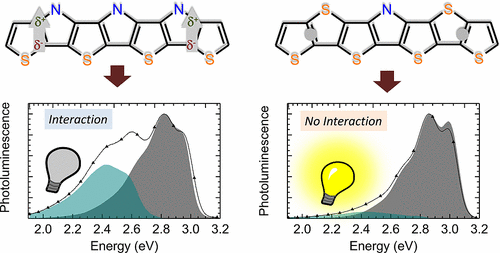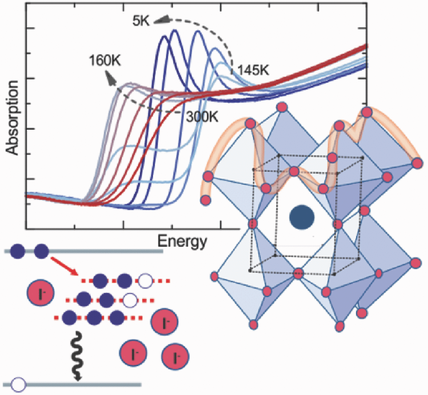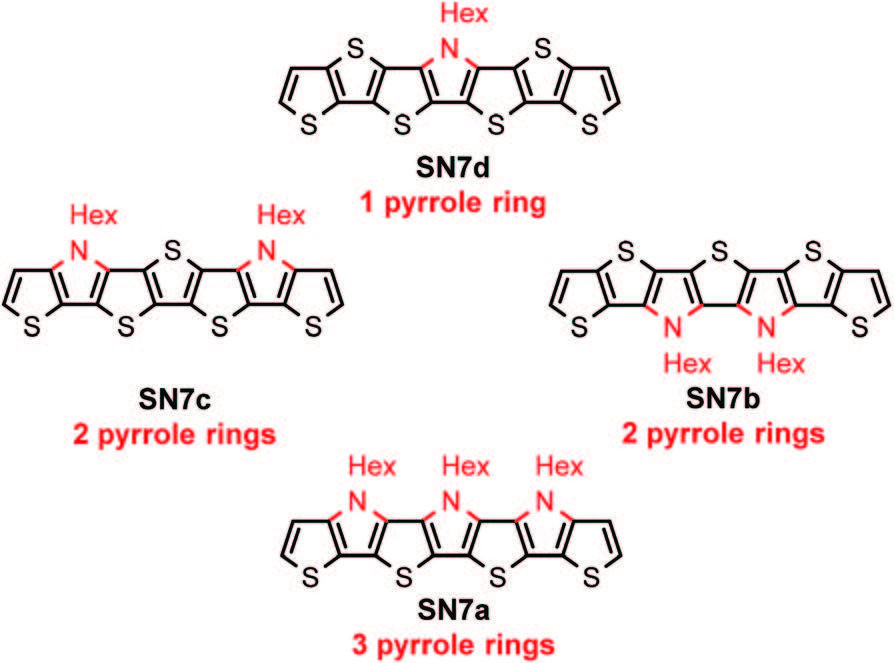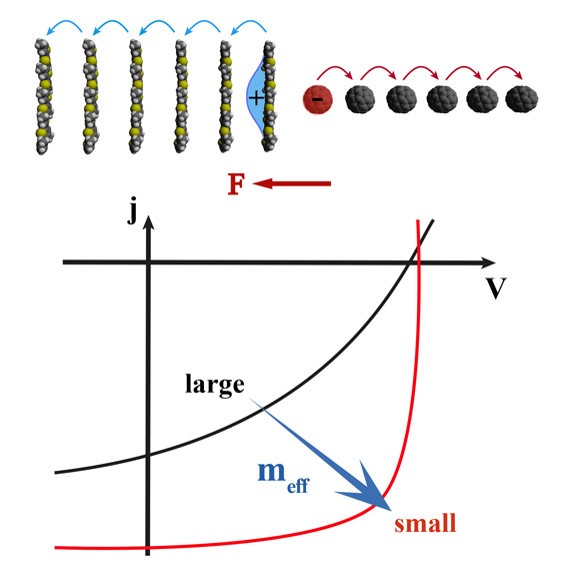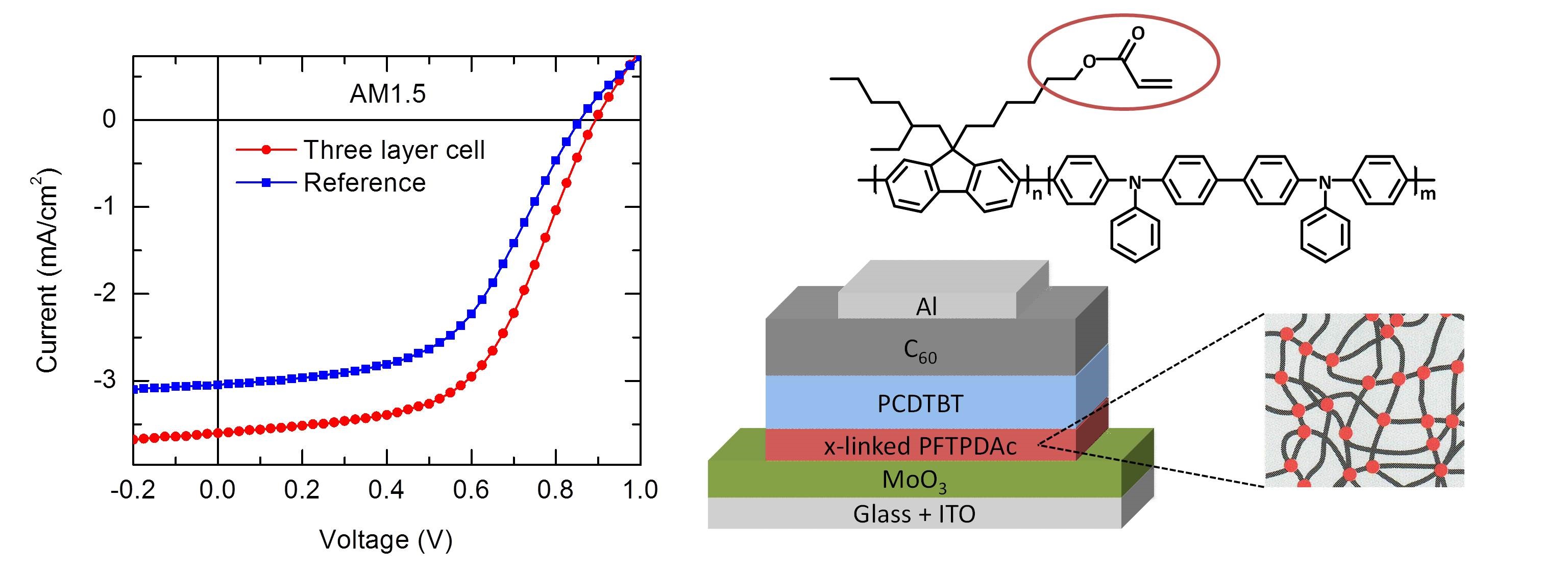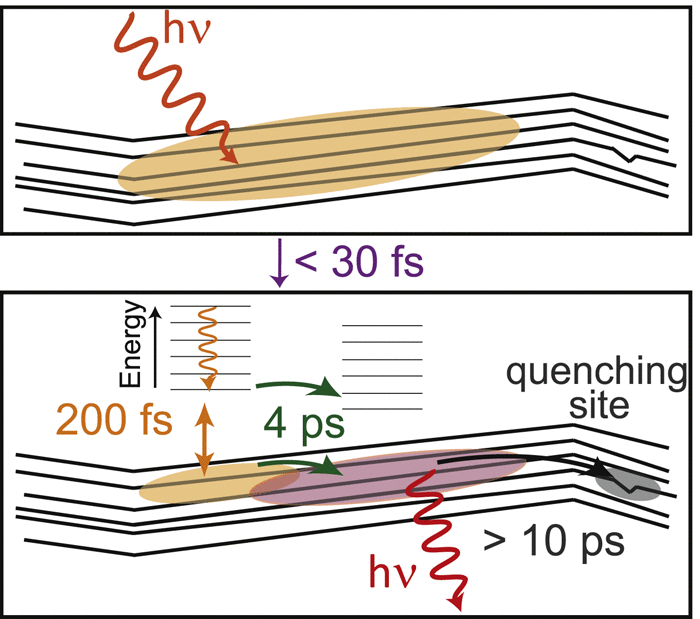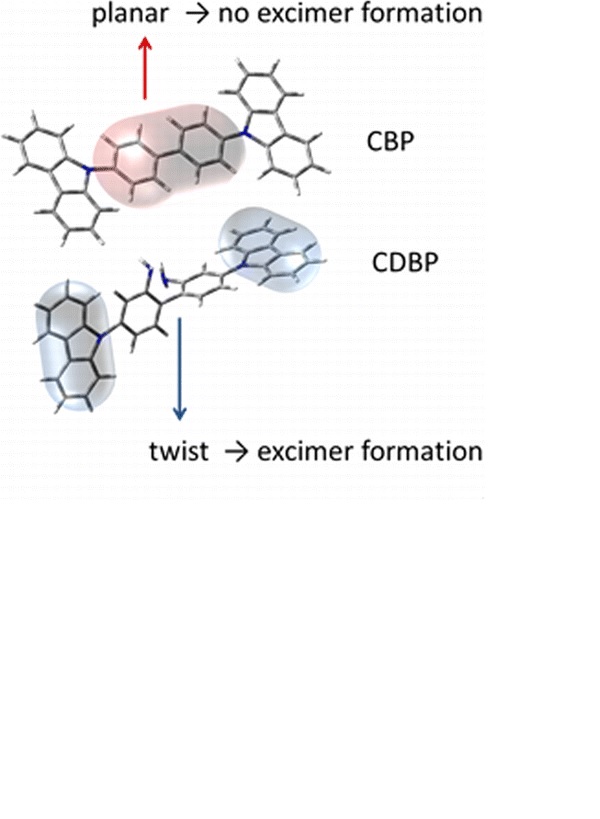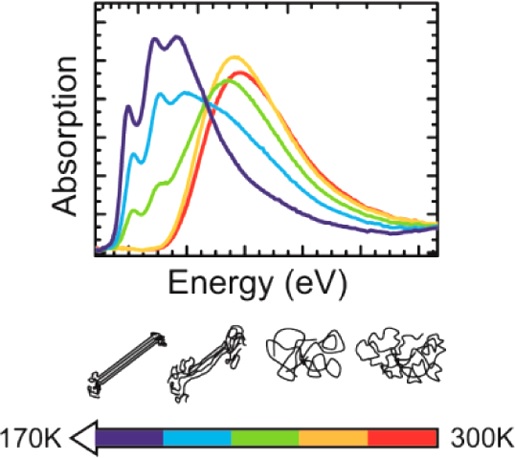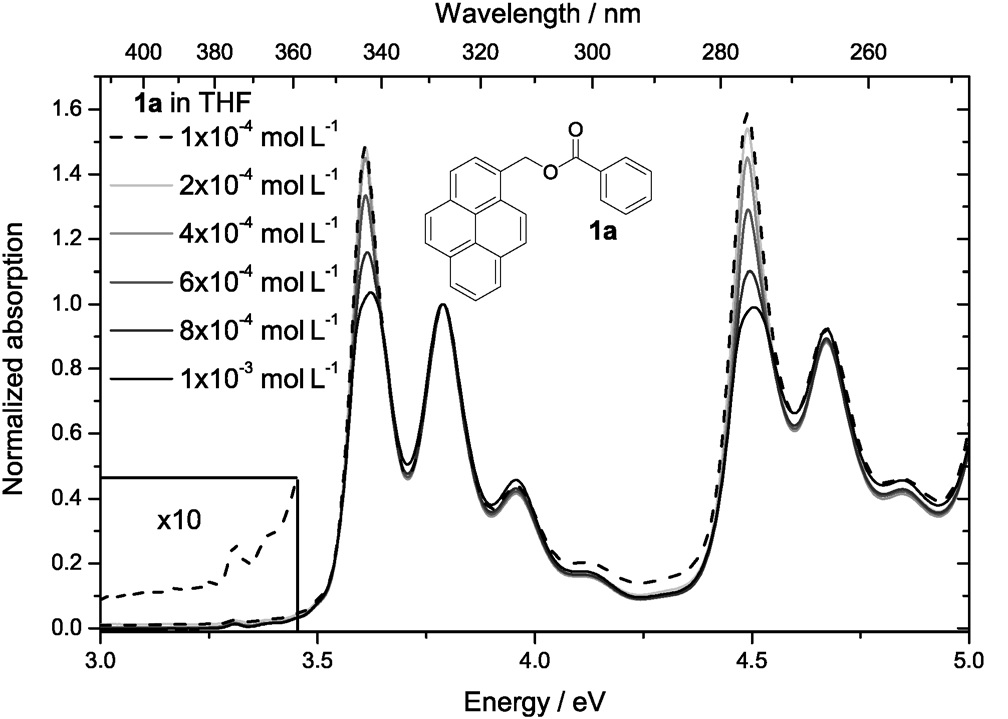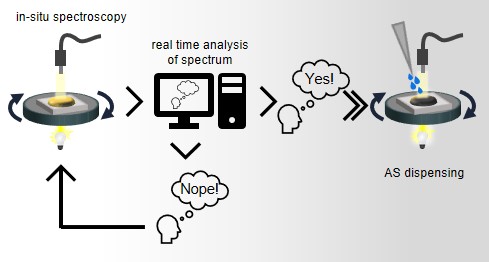
Efficient perovskite solar cells require metal halide perovskite (MHP) films of consistent and reproducible high quality. MHP films are frequently prepared through a solution-based solvent-engineering spin coating approach. This processing involves considering various controllable parameters (e.g. spin speed) and ones that are more difficult to control (e.g. changes in atmosphere) to fabricate MHP films reliably. To address this issue, we developed a closed-loop feedback system based on a multimodal optical in situ spectroscopy spin coater system. We combine this system with real-time monitoring and analysis of the optical spectra during the spin coating process. As soon as a parameter of interest reaches a predefined target level, perovskite crystallization is automatically induced by dispensing the antisolvent via a syringe pump. To demonstrate our approach, we optically monitor the precursor solution film thickness as the parameter of interest during the spin coating. We intentionally vary the evaporation kinetics by spin coating at different spin speeds between 2000 and 1250 rpm and compare our reactive method to the common time-based approach. We find that our method reliably counteracts effects like variation in solvent evaporation rate due to atmospheric changes and reduces the human impact on the processing, thus leading to reproducible film quality for all spin speeds without any optimization steps.